Research
Mass spectrometry is a fascinating analytical and biological technology. Our research interest is broad and we are working on organic, biological, environmental and forensic mass spectrometry. In Particular, our research focuses on electrochemical mass spectrometry.
The current projects are:
Develop novel quantitation methods for peptides without using standards by Coulometric Mass Spectrometry
Quantitation methods for peptides using mass spectrometry have advanced rapidly. These methods rely on using standard and/or isotope-labeled peptides, which might be difficult or expensive to synthesize. To tackle this challenge, we present a new approach for absolute quantitation without the use of standards or calibration curve based on coulometry combined with mass spectrometry (MS)1-4. In this approach, which we call coulometric mass spectrometry (CMS), the mass spectrum of a target peptide containing one or more tyrosine residues is recorded before and after undergoing electrochemical oxidation. We record the total integrated oxidation current from the electrochemical measurement, which according to the Faraday’s Law of coulometry, provides the number of moles of oxidized peptide. The ion intensity ratio of the target peptide before and after oxidation provides an excellent estimate of the fraction of the peptide that has been oxidized, from which the total amount of peptide is calculated. The striking strength of CMS is that it needs no standard peptide, but CMS does require the peptide to contain a known number of oxidizable groups. To illustrate the power of this method, we analyzed various tyrosine-containing peptides such as GGYR, DRVY, oxytocin, [Arg8]-vasotocin and angiotensinogen 1-14 with a quantification error ranging from -7.5% to +2.4 %. This approach is also applicable to quantifying phosphopeptides and could be useful in proteomics research. Recently, in collaboration with scientists at Merck Research Laboratories, we successfully applied our CMS method for quantifying N-nitrosamines with high sensitivity and selectivity, without use of any standard (JASMS, 2022, 33, 875-884). In addition, in collaboration with scientists at Johnson & Johnson and Merck Co., we successfully applied our CMS method for quantifying antibody deamidation degradation and host cell proteins (HCPs), without use of any standard (Anal. Chem., 2022, 94, 12490−12499). Therefore, peptides without oxidizable residues can be quantified after derivatization with an electrochemically active tag (Int. J. Mass Spectrom., 2024, 495, 117153).
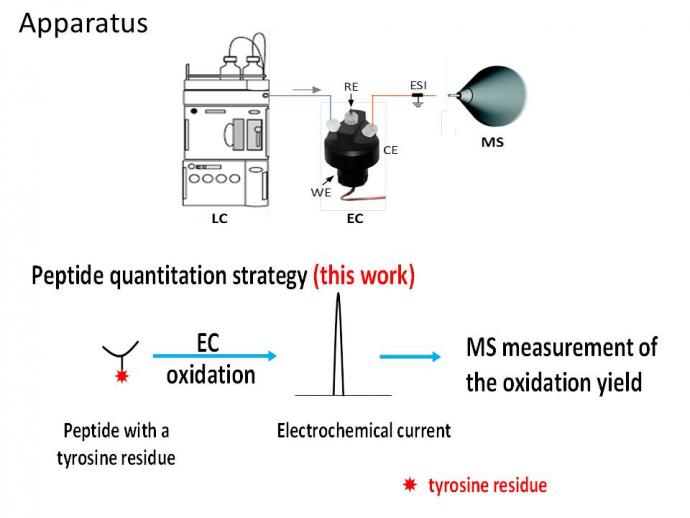
Scheme 1. Our LC/EC/MS apparatus and approach for quantitation of tyrosine-containing peptides.
Develop rapid and ultra sensitive method for detection of PFAS by high resolution MS
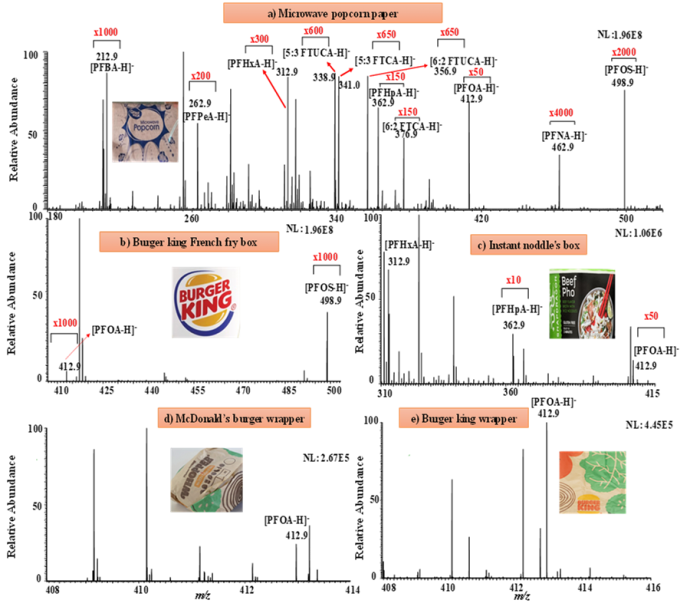
Traditional PFAS analysis by mass spectrometry (MS) is time-consuming, as laborious sample preparation (e.g., extraction and desalting) is necessary. Herein, we report fast detection of PFAS by paper spray (PS)-based MS techniques, which employs a triangular-shaped filter paper for sample loading and ionization (≤ 3 min per sample). In our study, PS-MS was first used for direct PFAS analysis of drinking water, tap water, and wastewater.(Journal of Hazardous Materials, 2023, Accepted) Interestingly, food package paper materials can be directly cut and examined with PS-MS for possible PFAS contamination (see Figure below). For samples containing salt matrices which would suppress PFAS ion signal, desalting paper spray mass spectrometry (DPS-MS), was shown to be capable of rapidly desalting, ionizing and detecting PFAS species such as per-flurooctanoic acid (PFOA) and per-flurosulphonic acid (PFOS). The retention of PFAS on paper substrate while salts being washed away by water is likely due to hydrophilic interaction between the PFAS polar head (e.g., carboxylic acid, sulfonic acid) with the polar filter paper cellulose surface. The DPS-MS method is highly sensitive (limits of detection:1.2-4.5 ppt) and can be applicable for directly analyzing soil extract and soil samples. These results suggest the high potential of PS-MS and the related DPS-MS technique in real-world environmental analysis of PFAS.
Probe Transient Electrochemical Intermediates by Mass Spectrometry
We have extended the desorption electrospray ionization (DESI),5 an ambient ionization method, to the direct analysis of liquid samples. Due to its direct sampling and ionization properties, this liquid sample DESI-MS (LS-DESI-MS)6 is versatile and has unique applications. For instance, it allows us to probe trasient electrochemical reaction intemediates7-9. Using a Waterwheel setup (Scheme 2), DESI-MS can be used to directly monitor electrochemical reactions from a rotating electrode surface.
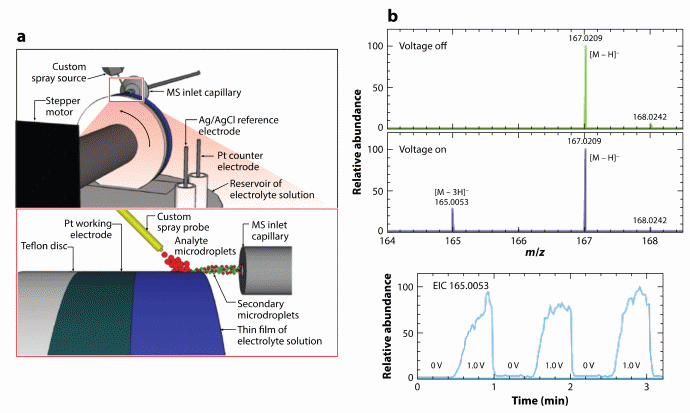
Scheme 2. Waterwheel setup allowing detection of transient electrochemical reaction intermediates from electrode surface by MS. Deprotonated diimine intermediate resulting from uric acid oxidaiton was detected at m/z 165, as shown in the right panel.
Furthermore, we applied electrochemistry to assist investigation of photoredox reaction mechanisms by MS.10
Electrochemical Reduction of Protein/Peptide Disulfide Bonds
Traditional methods for protein structural analysis involves chemical reduction of disulfide bonds prior to mass spectrometric detection, which is time-consuming. We have developed online electrochemical reduction approach11-14 which is reagentless and fast, facilitating MS elucidation of protein structures for proteomic research. For instance, after online electrochemical reduction of disulfide bonds, sequence coverage from MS/MS dissociation of resulting reduced protein ions (Scheme 3) are greatly improved.
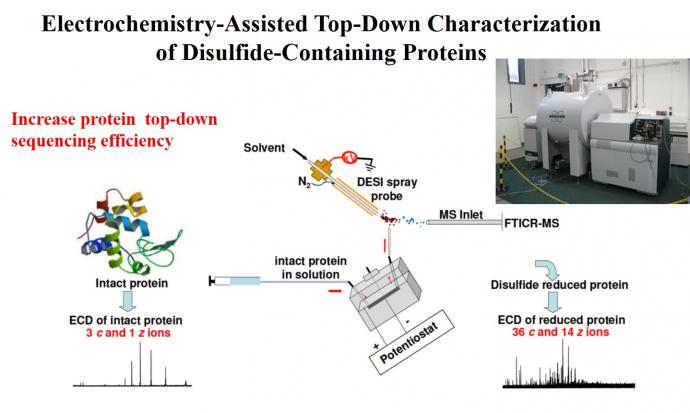
Scheme 3. Top-down characterization of proteins after online disuflide-bond reduction12
The online electrochemical reduction of disulfde bond can be also used to assist cross-linking MS to probe protein/protein interactions and protein 3D structures.15-16
Eluciation of Organometallic Reaction Mechanisms by in-situ Mass Spectrometry
Due to the direct sampling capability, we have applied liquid sample DESI-MS to detect Pd(0) species from Buchwald C-N coupling reaction.17
Furthermore, in collaboration with oragnic chemists, we have carried out extensive study for elucidating reaction mechanisms18-24, in particular, gold-mediated coupling reactions (Scheme 4), in which gold(III) complex were identified.
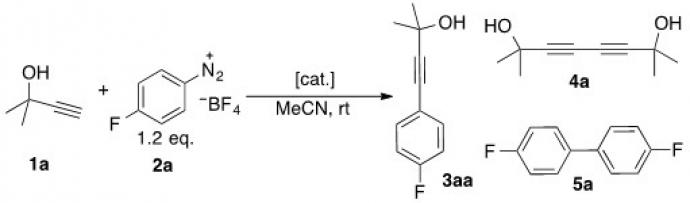
Scheme 4. Redox gold catalysis without using an external oxidant
Develop Atmospheric Pressure Ion Dissociation Methods
Tandem mass spectrometry is invaluable for chemical structure elucidation via examination of fragment ions after the activation and dissociation of gaseous precursor ions. However, neutral fragments are ignored and not detected by MS. Our lab has been working on atmospheric pressure thermal dissociation method (APTD)25-28 which dissociates ions outside mass spectrometer and thus allows re-ionization of neutral fragments. This approach helps to gain increased structure information about both proteins and organic molecules such as synthetic drugs. It could find applications in both forensic chemistry and proteomics.
Develop New Interfaces for Coupling LC with MS
LC/MS plays a significant role in analytical chemistry. However, high flow rate of eluents is not compatible with MS ionization. We have developed a novel splitting method29-31 which allows real-time MS monitoring of LC-separated analytes and subsequent online analyte collection. In this approach, a PEEK capillary tube with a micro-orifice drilled on the tube side wall is used to connect with LC column. A small portion of LC eluent emerging from the orifice can be directly ionized by LS-DESI-MS with negligible time delay (few ms) while the remaining analytes exiting the tube outlet can be collected following DESI-MS monitoring. The DESI-MS analysis of eluted compounds shows narrow peaks and high sensitivity, due to the extremely small dead volume of the orifice used for LC eluent splitting (as low as 4 nL) and the freedom to choose favorable DESI spray solvent. In addition, online derivatization using reactive DESI is possible for supercharging separated proteins and for enhancing their signals without introducing extra dead volume. Unlike UV detector used in traditional preparative LC experiments, our method is applicable to compounds without chromophores.
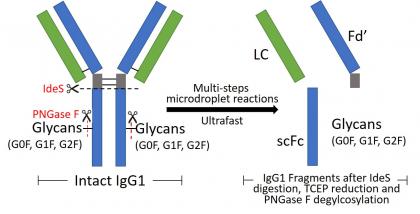
Microdroplet Ultrafast Reactions Speed Antibody Characterization
Recently, microdroplet reactions have aroused much interest because the microdroplet provides a unique medium where organic reactions could be accelerated by a factor of 103 or more. However, microdroplet reactions of proteins have been rarely studied. We reported the occurrence of multiple-step reactions of a large protein, specifically, the digestion, reduction, and deglycosylation of an intact antibody, which can take place in microseconds with high reaction yields in aqueous microdroplets at room temperature.32 As a result, fast structural characterization of a monoclonal antibody, essential for assessing its quality as a therapeutic drug, can be enabled. We found that the IgG1 antibody can be digested completely by the IdeS protease in aqueous microdroplets in 250 microseconds, a 7.5 millionfold improvement in speed in comparison to traditional digestion in bulk solution (>30 min). Strikingly, inclusion of the reductant tris(2-carboxyethyl)phosphine in the spray solution caused simultaneous antibody digestion and disulfide bond reduction. Digested and reduced antibody fragments were either collected or analyzed online by mass spectrometry. Further addition of PNGase F glycosylase into the spray solution led to antibody deglycosylation, thereby producing reduced and deglycosylated fragments of
analytical importance. In addition, glycated fragments of IgG1 derived from glucose modification were identified rapidly with this ultrafast digestion/reduction technique. We suggest that microdroplets can serve as powerful microreactors for both exploring largemolecule reactions and speeding their structural analyses. Now we could automate the sample injection and enable completing IgG digestion and analysis in 2 min for each sample (Anal. Chem., 2023, 95, 6, 3340–3348).
We also have strong interest in forensic and environmental applications of mass spectroemtry.
As the only forensic science undergraduate program in New Jersey, NJIT’s Forensic Science B.S. program provides theoretical foundation in the natural sciences and mathematics, as well as hands-on training in field methods, analytical skills and laboratory procedures currently used by forensic scientists in crime and investigative laboratories at the state and federal level. A new forensic science laboratory has been renovated.

Our Industrial Collaborators:



![]()
References
2. P. Zhao, Y. Guo*, H. D. Dewald*, H. Chen*, Int. J. Mass Spectrom. 2019, 443, 41-45.
3. C. Xu, Q. Zheng, P. Zhao, J. Paterson, H. Chen*, J. Am. Soc. Mass Spectrom. 2019, 30, 685–693.
5. Takats, Z.; Wiseman, J. M.; Gologan, B.; Cooks, R. G*. Science, 2004, 306, 471.
6. Miao, Z.; Chen, H*. J. Am. Soc. Mass Spectrom. 2009, 20, 10.
7. T. A. Brown, H. Chen*, R. N. Zare*, Angew. Chem. Int. Ed., 2015, 54, 11183 –11185
8. T. A. Brown, H. Chen*, R. N. Zare, J. Am. Chem. Soc. 2015, 137, 7274.
9. T. A. Brown, N. Hosseinei-Nassab, H. Chen*, R. N. Zare*, Chemical Science, 2016, 7, 329.
10. Y. Cai, J. Wang, Y. Zhang, Z. Li, D. Hu, N. Zheng*, H. Chen*, J. Am. Chem. Soc., 2017, 139, 12259.
11. Li, J.; Dewald, H. D.; Chen, H*. Anal. Chem., 2009, 81, 9716.
12. Y. Zhang, W. Cui, H. Zhang, H. D. Dewald, H. Chen*, Anal. Chem. 2012, 84, 3838.
13. Y. Zhang, H. D. Dewald, H. Chen*, J. Proteome Res. 2011, 10, 1293.
14. Q. Zheng, H. Zhang*, H. Chen*, Int. J. Mass Spectrom. 2013, 353, 84.
15. Q. Zheng, H. Zhang, S. Wu, H. Chen*, J. Am. Soc. Mass Spectrom. 2016, 27, 864-875.
16. Q. Zheng, H. Zhang*, L. Tong, S. Wu, H. Chen*, Anal. Chem. 2014, 86, 8983-8991
17. Q. Zheng, Y. Liu*, Q. Chen*, M. Hu, R. Helmy, E. C. Sherer, C. J. Welch, H. Chen*, J. Am. Chem. Soc., 2015, 137, 14035-14038
18. R. Cai, M. Lu, E. Aguilera, Y. Xi, N. Akhmedov, J. Petersen, H. Chen*, X. Shi*, Angew. Chem. Int. Ed., 2015, 54, 8772 –8776
19. H. Peng, R. Cai, C. Xu, H. Chen*, X. Shi*, Chemical Science, 2016, 7, 6190–6196.
20. J. Wang, S. Zhang, C. Xu, L. Wojtas, N. G. Akhmedov, H. Chen, X. Shi,* Angew. Chem. Int. Ed., 2018, 57, 6915 –6920.
21. M. Lu, Y. Su, P. Zhao, X. Ye, Y. Cai, X. Shi,* E. Masson,* F. Li, J. L. Campbell, H. Chen*, Chem. – Eur. J., 2018, 24, 2144-2150
22. X. Ye, P. Zhao, S. Zhang, Y. Zhang, Q. Wang, C. Shan, L. Wojtas, H. Guo*, H. Chen*, X. Shi,* Angew. Chem. Int. Ed., 2019, 58, 17226 –17230.
23. J. Wang, C. Wei, X. Li, P. Zhao, C. Shan, L. Wojtas, H. Chen, X. Shi*, Chem. Eur. J. 2020, 26, 5946-5950.
24. T. Yuan, P. Zhao, S. Teng, Y. Yi, J. Wang, C. Shan, L. Wojtas, J. Jean, H. Chen*, X. Shi*, Chem, 2020, 6, 1420–1431.
25. H. Chen, L. S. Eberlin, R. G. Cooks*, J. Am. Chem. Soc. 2007, 129, 5880.
26. H. Chen, L. S. Eberlin, M. Nefliu, R. Augusti, R. G. Cooks*, Angew. Chem. Int. Ed., 2008, 47, 3422.
27. P. Liu, P. Zhao, R. G. Cooks*, H. Chen*, Chemical Science, 2017, 8, 6499.
28. P. Zhao, T. White, R. G. Cooks,* Q. Chen,* Y. Liu, H. Chen*, J. Am. Soc. Mass Spectrom. 2018, 29, 2317–2326.
29. Y. Cai, D. Adams, H. Chen*, J. Am. Soc. Mass Spectrom. 2014, 25, 286.
30. Y. Cai, Y. Liu, R. Hemly, H. Chen*, J. Am. Soc. Mass Spectrom. 2014, 25, 1820.
31. Y. Cai, Q. Zheng, Y. Liu, R. Helmy, J. A. Loo, H. Chen*, Eur. J. Mass Spectrom. 2015, 21, 341–351.